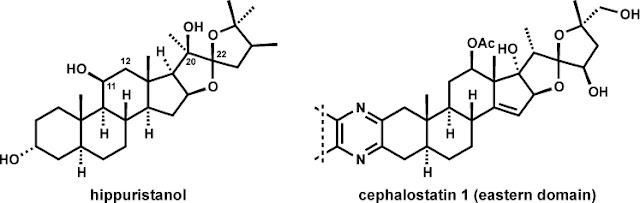
Hippuristanol is
an antiproliferative agent that selectively targets an RNA helicase initiation
factor [(eIF)4A] to block the translation stage of eukaryotic protein
biosynthesis. This binding interaction triggers potent cytotoxic activity
against cultured tumor cells and establishes RNA helicases as novel anticancer
targets. Structurally, hippuristanol is a polyoxygenated steroid with a C22
spiroketal unit in a thermodynamically unfavorable stereochemical configuration
(vida infra). The steroid framework is
also hydroxylated on the beta-face at C11 and C20, that latter being a
quaternary stereogenic position. Hippuristanol shares certain topological
structural features with the ‘eastern’ substructure of cephalostatin 1, a
dimeric steroidal pyrazine with uniquely selective and exquisitely potent
anticancer properties. Pursuit of the currently unknown cellular target and
mechanism of action of cephalostatin 1 is the subject of extensive research
efforts in a number of academic laboratories and institutions.

A concise
partial synthesis of hippuristanol, disclosed in 2010 from the laboratory of
Pierre Deslongchamps, starts from hecogenin acetate, a plant-derived bulk
chemical with side chain spiroketal functionality that can be rearranged and
elaborated as needed. However, in order to make use of this inexpensive sapogenin as a precursor to hippuristanol, hecogenin’s C12 oxo moiety must be
transposed to the C11 position. With this objective in mind, Deslongchamps and
co-workers first employ osmium tetroxide-catalyzed dihydroxylation of the silyl
enol ether derived from hecogenin to accomplish net alpha-hydroxylation at C11.
The ketol thus obtained is then subjected to treatment with alkali under
thermodynamically forcing conditions and rearrangement to the more stable ketol
ensues, presumably via the intermediacy of the bracketed enediol shown above.
This methodology was developed in the 1990’s by industrial chemists at Pfizer
for the production of a steroidal cholesterol absorption inhibitor. The
11-keto-12beta-acetoxy intermediate then undergoes reduction with calcium
metal, which effectively shifts the C12 keto group of hecogenin to C11. This
reduction probably involves an initial epimerization of the C12 position to
place the acetate in an axial position which would mechanistically facilitate
the requisite 1,2-elimination.

In order to
secure the C20 tertiary alcohol stereocenter, hecogenin’s C22 spiroketal must
be truncated to a 20-oxo-pregnane. Historically, this has been achieved by
implementation of Russell Marker’s degradation protocol, a three-step route
that efficiently provides 3-hydroxypregna-5,16-dien-20-ones
from various plant-derived sterols. The classical Marker degradation involves a
base-mediated elimination (step 3) to install a delta(16,17) double bond. This
results in the loss of stereochemical information (two stereocenters are
abolished) and oxygenation at C16. In order to retain the beta-configuration at
carbons 16-17 and retain the C16 hydroxyl, Deslongchamps has modifed step 3 of
the Marker degradation by treating the intermediate derived from the chromic
acid oxidation with vinylmagnesium bromide. This delivers an allylic alcohol
that can then be oxidatively cleaved to provide the requisite beta-hydroxy
ketone needed for elaboration to the natural product. Deslongchamps’
modification of the Marker degradation may be a generally useful process for
the preparation of complex oxygenated steroids. In the next step, addition of a
lithiated alkyne to the C17 methyl ketone stereoselectively affords the product
of chelation control, the C20beta-tertiary alcohol.

Without
question, the most interesting transformation in the synthesis of hippuristanol
is the mercury(II)-catalyzed spiroketalization that generates semiprotected
22-epi-hippuristanol in one step from a 3-alkyn-1,7-diol precursor. A proposed
mechanism was advanced by the authors involving intramolecular 5-exo-dig oxymercuration
of the 16-hydroxyl onto the triple bond. A subsequent ketalization event
establishes the C22 spirocarbon center. This latter mechanistic aspect is of
great interest, as the reaction stereoselectively produces only one of two
possible diastereomeric products (22S/22R = 99.9:0.1). Evidence is provided to suggest that
this is a thermodynamically controlled process: treatment of hippuristanol
under the aqueous mercury(II) reaction conditions leads to complete
isomerization of the natural product to 22-epi-hippuristanol. Indeed, upon
inspection of the possible diastereomeric products (stereo structures shown
above), one can see that the observed product is stabilized by two anomeric effects and that hippuristanol’s bicyclic spiroketal is in the non thermodymanically-favored
configuration (due to one anomeric effect). Therefore, the stereochemical
outcome of the reaction is probably due to equilibration rather than selective
kinetic ketalization of the nucleophilic hydroxyl onto the intermediate oxonium
species from the alpha-face. Debenzylation and spiroketal isomerization
completed the expedient synthesis of hippuristanol. The observation that
partial epimerization of C22 to the natural configuration can only be
accomplished in an aprotic acidic
system implies the existence of an intramolecular hydrogen bond between the
hippuristanol’s C20 tertiary hydroxyl group and a spiroketal oxygen atom.





