

The method reported by the
laboratory of John Groves at Princeton University is a manganese-catalyzed
process that uses sodium azide as the azide source. Mechanistically, this
chemistry is analogous to Groves’ previously disclosed manganese-catalyzed C-H
fluorination wherein a fluoride ligand axial to manganese transfers
fluorine to a carbon-centered radical derived from the hydrocarbon substrate.
For azidation, the fluoride is simply replaced by an azide source (NaN3).
The substrate radical is then trapped by the in situ-generated Mn(IV)-azide
complex to construct the carbon-nitrogen bond. Both manganese porphyrins as
well as Mn salen-type Jacobsen catalysts are competent participants in the
catalytic cycle and the novel azidation protocol can be run under air. Under
the optimized reaction conditions, estrone acetate is converted predominantly
to a C9-a-azide. A diazidation product
wherein both benzylic positions (C6 and C9) are functionalized is also observed
as a major side product.
John Hartwig’s group has also
developed a method for late-stage azidation of tertiary and benzylic C-H bonds
using an iron catalyst and Zhdankin’s hypervalent azidoiodinane reagent. For a
nice introductory overview of Hartwig’s technology, see this blog post.
Azidation of TBS-protected estrone, under the iron-catalyzed conditions,
furnishes the corresponding C6-a-azide
in modest yield. The stereodivergent nature of the two related azide-forming processes
is somewhat striking.
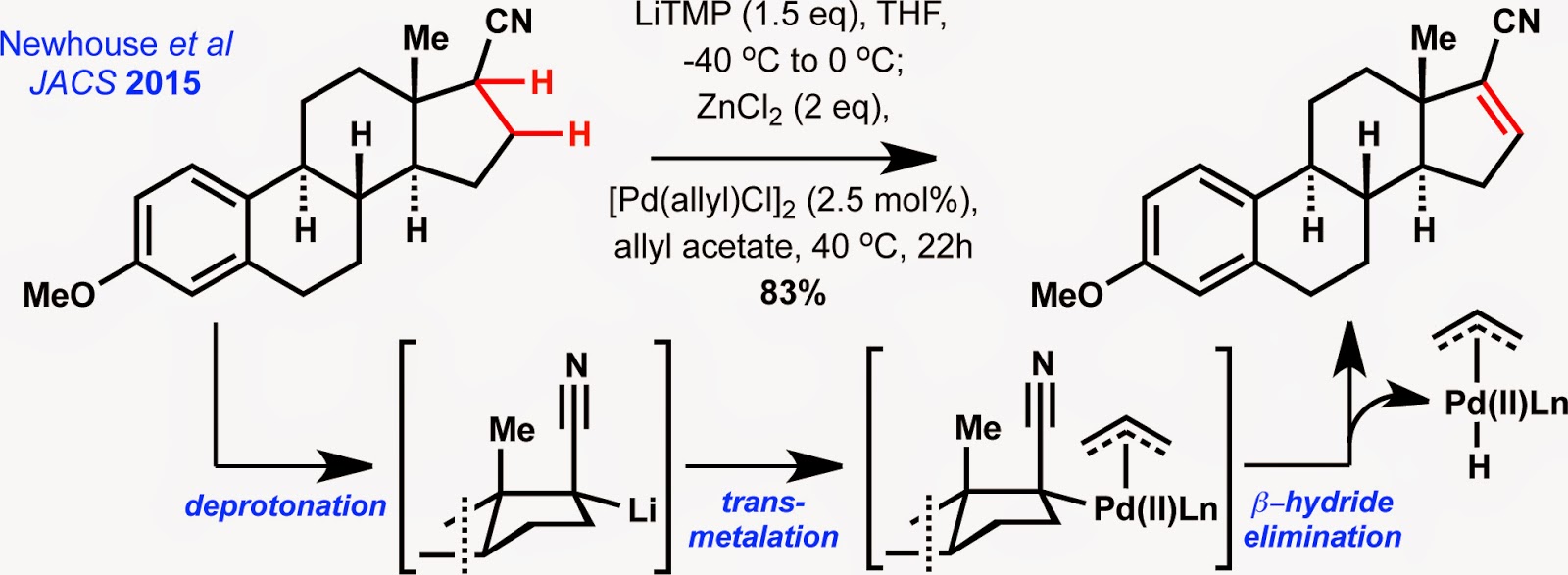
On a somewhat unrelated note, the
laboratory of Timothy Newhouse at Yale University has disclosed a new
palladium-catalyzed methodology for a,b-dehydrogenation
of esters and nitriles. The method nicely complements (and, in some cases, exceeds)
earlier, more classical approaches to achieve a,b-unsaturation
such as the Saegusa-Ito oxidation or the Sharpless selenoxide elimination (later extended by Grieco). The novel
reaction from the Yale group is compatible with nitrile substrates derived from
estrone (shown above), cholesterol and androstenedione-type steroids.
No comments:
Post a Comment