Estrogen deficiency in the human
brain is observed as a rather serious side effect of preventative oophorectomy,
the surgical removal of ovaries, which is increasingly performed today in
gynecological oncology. The deficiency arises because women who have had
bilateral oophorectomy surgeries lose most of their ability to produce the
hormones estrogen and progesterone. The sudden inability to produce estrogen
initiates what is referred to as ‘surgical menopause,’ which is generally
accompanied by an abrupt onset of neurological and psychiatric maladies
triggered by the hormonal deficiencies. Those menopausal indications are
commonly addressed through equine estrogen-based hormone replacement therapy. Unfortunately,
hormone replacement therapy is not desirable for all symptomatic women due to
the peripheral side effects and tumor-promoting properties of estrogen.
Therefore, the invention and commercialization of a safe and effective
treatment of the adverse consequences of estrogen deficiency in the brain, which
include depression and impaired sexuality, is a major unmet medical need.

The development of brain-selective
estrogen therapies has been a formidable medical challenge. Recent efforts to generate
neuroselective estrogen receptor modulators have been achieved through GLP-1
receptor-mediated cellular targeting and intracellular delivery. This strategy
uses a covalently attached peptide carrier, the glucagon-like peptide-1
(GLP-1), that delivers estrogen selectively to specific tissues in order to
improve the therapeutic index of estrogen. Tissue specific delivery of estrogen
is limited to cells that co-express both estrogen as well as GLP-1 receptors. The
(GLP-1)-estrogen conjugates (Figure above, lower panel) that were discovered contain
plasma-stable linkages and are reported to exhibit improved sex-independent
efficacy over either of the individual hormones alone for the treatment of
diabetes and obesity.
In order to develop an orally
bioavailable treatment option for brain-selective estrogen therapy, an
alternate small-molecule strategy is desired. A research group led by Laszlo Prokai has initiated preclinical evaluation of a unique synthetic steroid that was designed for the treatment of estrogen-responsive central disorders. His
approach involves development of a ‘bioprecursor prodrug’ that undergoes
enzymatic bioactivation to 17b-estradiol
(E2) by a reductive process catalyzed by an enzyme that is selectively
expressed in the brain. The specific bioprecursor, 10b,17b-dihydroxyestra-1,4-dien-3-one
(or DHED), unlike a conventional prodrug, does not contain any auxiliary promoieties
that require enzymatic or chemical cleavage. Instead, a short-chain
NADPH-dependent reductase (SDR) promotes the reductive bioactivation of DHED
through hydride transfer from the coenzyme NADPH (mechanism shown above, top
panel) to the C1 position of the A-ring dienone followed by elimination of
water to furnish E2. In vitro metabolism studies using tissue
homogenates indicated that DHED was converted to E2 in the brain,
but not in peripheral tissues such as the uterus. In vivo experiments using
deuterated-DHED demonstrated that d3-E2
was produced exclusively in the brain. Estrogen was not detected in peripheral
tissues, nor was DHED detected in the brain. Oral, intravenous and subcutaneous
administration of the prodrug to ovariectomized rodents resulted in rapid,
brain-selective bioconversion to E2.

In order to evaluate DHED in a
preclinical model of an estrogen-responsive human CNS disorder, a Forced Swim
Test (FST) study was conducted. The Porsolt Forced Swim Test is a widely accepted
animal model of the human condition of depression, used to screen for
antidepressant-like pharmacological activity. The test is centered on a
rodent’s response to the threat of drowning and the result of the test, a quantitation
of reduced behavioral immobility, is interpreted as measuring susceptibility to
negative mood. In an FST study, animals are subjected to two trials during
which they are forced to swim in a glass cylinder filled with water, from which
they cannot escape. The second trial is performed 24 hours after the first. The
time that the animal spends in the second trial without making movements beyond
those required to keep its head above water, referred to as immobility time, is
measured and is known to be decreased by antidepressant drugs. The FST is also
referred to as the “behavioral despair test.”
 |
| Reproduced from: Science Translational Medicine 2015, 7, pp. 297ra113. |
When subcutaneously administered to
ovariectomized rodents at identical doses, DHED treatments engendered decreased
FST immobility times as compared to direct administration of E2 (FST
data shown above, reproduced from Science Translational Medicine 2015, 7, pp. 297ra113). Importantly, co-injection of a
high-affinity estrogen receptor antagonist (ICI 182,780, structure depicted
above) blocked the antidepressant-like effect in both treatment groups,
suggestive of an estrogen receptor-mediated mechanism of action. The profoundly
reduced behavioral immobility induced by DHED in the FST suggests that this
unique bioprecursor prodrug holds promise as a potentially safe therapy to
alleviate hypoestrogenic depression resulting from surgical menopause. The
treatment should be devoid of adverse peripheral side effects associated with
the use of systemic estrogens. Moreover, the physicochemical properties of DHED,
such as lipophilicity and intrinsic aqueous solubility, are significantly
improved relative to the corresponding properties of 17b-estradiol. The attractive biopharmaceutical properties of the
small-molecule DHED, in comparison to those of E2 or estrogen-peptide
conjugates, could facilitate applications involving oral administration, which
is a much-coveted feature of a drug candidate.
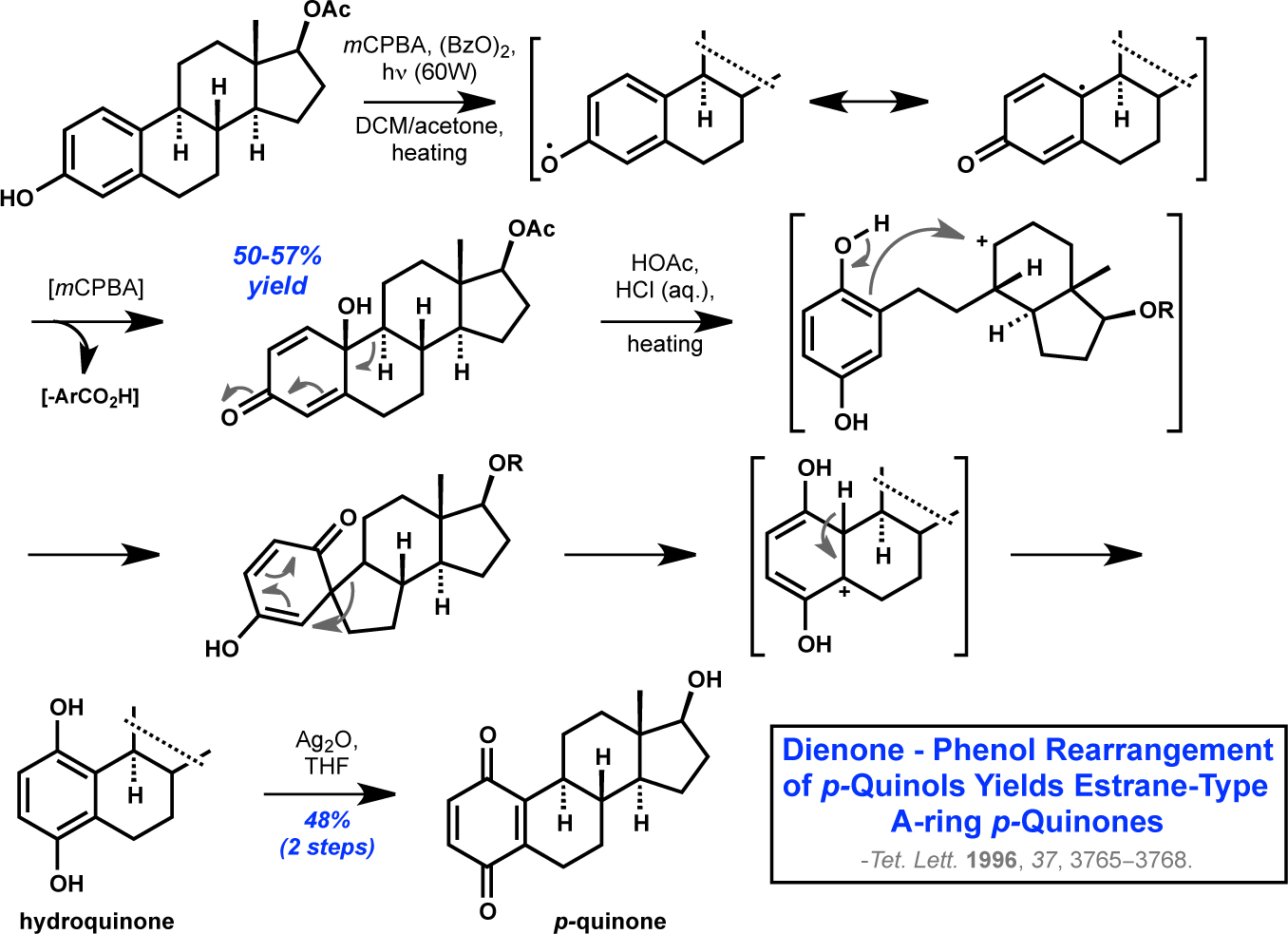
DHED is a synthetic molecule
obtained from chemical oxidation of estrogen derivatives. Upon exposure of an
estrogen to an initiator (benzoyl peroxide) and an oxidant (mCPBA) with simultaneous irradiation
from a 60-Watt light bulb, the A-ring phenol is converted to its corresponding p-quinol in moderate yield via the
radical mechanism depicted in the scheme above. It’s interesting to note that
subsequent treatment of quinols related to DHED with a strong Brönsted acid under thermally
forcing conditions promotes a fascinating skeletal rearrangement reaction that ultimately
yields an A-ring quinone through the intermediacy of its corresponding
hydroquinone. Estrane-type A-ring p-quinones have been reported to exhibit moderate cytotoxicity against certain cell lines. It remains to be seen as to whether or not this unique mode of
reactivity will preclude the use of DHED in applications involving intestinal
drug absorption from the gut, given that gastric pH is, of course, strongly acidic.
For women, estrogen deficiency is a very serious illness. If not treated in time it may also lead to a series of complications. E.g. cancer. as for the treatment of this type of disease, personally think that cancer biomarkermay play a role
ReplyDelete