
In a previous series of posts at
this site, we highlighted a placebo-controlled human study with purified
limonin glucoside (LG) in which significant health benefits were observed. The
disclosure of these exciting results by the U.S. Department of Agriculture (USDA) provoked a thought exercise
concerning the downstream supply chain issues that this development program
will likely face. The USDA team led by Darshan Kelley was able to obtain about
100 grams of high-purity LG through development and execution of an optimized
extraction process starting from molasses, a by-product obtained from citrus
processing waste streams. However, in order to conduct additional clinical and
toxicological testing of this unique limonoid natural product, multiple
hundreds of grams (and likely kilograms) of API will be required at various stages. Given the
architectural complexity of limonin and related substances derived thereof,
does partial or total chemical synthesis provide a viable sourcing alternative?
In this post, we will examine the recently disclosed first total synthesis of
racemic limonin and compare the productivity of this process with that of the
optimized isolation from citrus molasses that was previously developed by the
USDA.
We should begin by noting that the
completion of the first total synthesis of limonin is an exceptional
accomplishment, given that this limonoid has been known as the bitter principle
derived from citrus for over 150 years. Limonin, an oxidatively fragmented
17-furylandrostane, is a highly complex small molecule, on par with other
notable limonoid family members including azadirachtin and libiguin A. The
detailed molecular structure of limonin remained unknown until 1960, when a
determination was finally made using chemical derivatization and X-ray
diffraction methods. More than fifty years later, in 2015, we now can begin to
discuss the state-of-the-art laboratory methods that enabled the first and only
chemical synthesis of limonin. The total synthesis of limonin, reported by the
Japanese team led by Shuji Yamashita and Masahiro Hirama, can roughly be broken
down into two synthetic “phases,” as outlined in the Figure above. In phase 1,
the 4,4,8-trimethylandrostane steroidal framework is constructed from readily
available geraniol. The critical synthetic operations that led to the
successful execution of phase 1 include a tandem radical polycyclization using Barry
Snider’s manganese(III)-mediated chemistry, followed by a standard Robinson
annulation to forge the steroid A-ring. This portion of the route provides
access to the requisite androstane system containing the appropriate
methylation and oxidation patterns for the completion of the target structure.
However, 17 synthetic steps are required to produce the desired intermediate
for initiation of phase 2 of the route, oxidative fragmentation and elaboration
of the eastern D-ring and western A-ring. The identification of an abundant,
plant-derived sterol that contains oxygenated functionality at carbon positions
3, 7 and 19, to be used as a starting material for a semisynthetic process, would
significantly improve the practicality of the overall approach by reducing the step
count. Phase 2 of the Tohoku University process is truly innovative and
provides a pioneering blueprint for late-stage synthetic manipulations
involving complex limonoid systems.

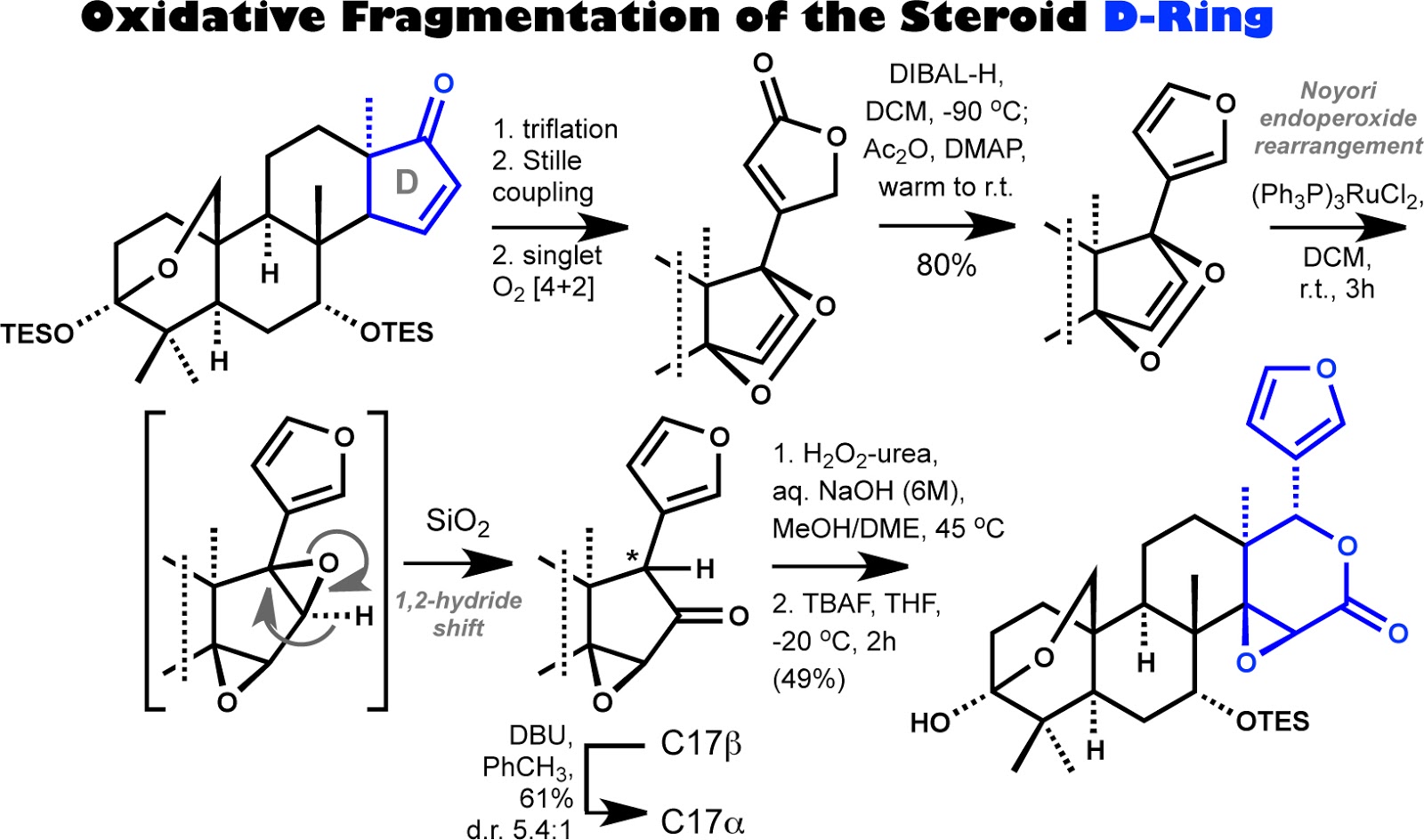
The portion of the inaugural limonin
synthesis that is truly worthy of highlighting is the ‘phase 2’ oxidative
fragmentation and elaboration of the androstane D-ring, a 13a-cyclopentenone, into the intact eastern
limonin substructure. The synthetic technology depicted in the Scheme above
will undoubtedly guide and enable future synthetic studies targeting limonoid
systems that were previously considered synthetically inaccessible due to
excessive chemical complexity. The overall conversion is initiated by
installation of a butenolide system via a Stille coupling protocol that has
been used previously to synthesize cardenolide natural products. Singlet oxygen
[4+2] cycloaddition then simultaneously functionalizes the steroidal C13 and
C17 positions in a single operation. As originally reported by Karel Wiesner in
the 1980s, the butenolide is a latent synthetic form of a furan ring system and,
notably, this reductive conversion could be accomplished without harming the
sensitive endoperoxide. Fortunately, upon exposure to a ruthenium(II) catalyst
as described by the Nobel laureate Noyori in the late 1980s, the endoperoxide functionality
was successfully fragmented to an oxy radical intermediate that underwent
further conversion to a bis-epoxide. Then, upon exposure of the intermediary
bis-epoxide to mildly acidic silica gel, a suprafacial 1,2-hydride shift
ensued, giving rise to an epoxy ketone albeit with the undesired stereochemical
configuration at C17. Base-mediated epimerization of the C17 position followed
by a Baeyer-Villiger ring-expansion of the five-membered D-ring finally
completed the stereocontrolled construction of a fully elaborated limonin
eastern substructure, suitably functionalized for late-stage oxidative manipulation
of the A-ring.

So how does this work impact the
supply chain question that was advanced at the top of this post? As I have
stated elsewhere, the metric for the degree of success of a synthetic project
can be quantified by answering the question: ‘How does this
technology compare with extraction from natural sources or microbial production
by an optimized fermentation process?’ Academic synthetic work can be both
virtuosic in its educational value for students, and, at the same time,
completely impractical as judged by industrial standards. The best synthetic
work is academically and aesthetically satisfying, but also useful in the lab
on a meaningful scale. In the case of limonin, similar to the infamous example provided
by azadirachtin, the state-of-the-art in synthetic chemical methodology falls
short when compared with isolation of drug substance from natural sources. As
noted previously, using the optimized USDA extraction process and estimating
conservatively, approximately two liters of citrus molasses can be processed in
four to six weeks to produce about 18 grams of high-purity limonin glucoside,
suitable for human studies. Moreover, the molasses feedstock is a worthless
by-product of citrus processing that juice producers are happy to dispose of.
On the other hand, the chemical synthesis of limonin in racemic form, in all likelihood,
required several man-years of effort in order to produce less than one
milligram. This comparison is, admittedly, harsh and perhaps unfair, given that
the two research groups involved in the work obviously had entirely different research
goals. However, the comparison is nonetheless instructive and should be taken
into consideration before it is asserted that organic chemistry has ‘matured,’
and that any molecule can be synthesized, given the appropriate time and resources.
The challenge presented by a chemical synthesis of limonin disputes that fundamentally flawed notion.
No comments:
Post a Comment